REPORTE DE CASOS CLÍNICOS
Enfermedad de Wilson: forma nueropsiquiátrica dominante presentación de un caso y su interpretación fisiopatológica basada en resonancia magnética del encéfalo
Marco A. Castañeda1; Rodrigo Ubilluz2; Carmen Ávalos3; Diego Escalante4; Juana Nicoll5.
1 Profesor Asociado de Medicina, Facultad de Medicina, UNMSM.
2 Profesor Principal de Medicina, Facultad de Medicina, UNMSM-USMP.
3 Profesora Invitada, Facultad de Medicina, UNMSM - UPCH.
4 Residente de Radiología, Facultad de Medicina, UPCH.
5 Estudiante de Medicina, Universidad Particular "Ricardo Palma ".
RESUMEN
Se presenta un caso clínico de la enfermedad de Wilson. Una mujer de 26
años de edad, comenzó a mostrar trastornos psíquicos, a los que se
añadieron signos neurológicos, tales como temblor asimétrico de las
manos, parkinsonismo, distonía; últimamente, presentó mutismo y
disfagia. La exploración oftalmológica demostró la presencia del anillo
de Kayser - Fleischer en la membrana de Descemet. Se comprobó alteración
del metabolismo del cobre en cuanto a reducción de la ceruloplasmina
sérica y aumento de la excreción urinaria de cobre. Mediante
laparoscopía y biopsia se puso en evidencia cirrosis hepática.
La investigación de las estructuras del encéfalo con resonancia
magnética (IRM) reveló atrofia frontotemporal del cerebro, y proceso
degenerativo de los ganglios basales, el cerebelo y el tronco
encefálico, datos que pueden ser utilizados para sugerir la probable
fisiopatología neuropsíquiátrica. La asociación con membrana
cricofaríngea causante de estenosis y disfagia cervical intensa no ha
sido mencionada anteriormente.
Palabras claves: Enfermedad de Wilson, Forma Neuropsiquiátrica Cirrosis.
SUMMARY
This is a presentation of a clinical case of Wilson's disease. The patient is a 26 year old woman who began to evidence psychological symptoms, which were later accompanied by neurological manifestations such as asymmetrical hand tremor, parkinsonism, dystonia and later on, dysphagia and mutism. The ophtalmological examination found a Kayser - Fleischer ring in Descemet's membrane. There was disturbance of copper metabolism documented with reduction of serum ceruloplasmin and increase of the urinary excretion of copper. Cirrhosis was demonstrated through laparoscopy and liver biopsy. The brain magnetic resonance showed frontotemporal atrophy and a degenerative process at the basal ganglia, cerebellum and brain stem. This information could suggest probable neuropsychiatric physiopathology. The stenosis and intense cervical dysphagia, associated with the crycopharyngeal membrane, has not been mentioned previously.
Key Words: Wilson's Disease, Neuropsychiatric type Cirrhosis.
INTRODUCCIÓN
La enfermedad de Wilson es representativa de una categoría nosológica introducida hace un siglo por científicos perspicaces que utilizaron los incipientes recursos técnicos disponibles en aquella época auroral. Se ha establecido desde entonces que el mecanismo patogénico primario es el defecto hereditario, autosómico y recesivo, localizado en el gen ATP7B del cromosoma l3; la consecuencia metabólica es la acumulación de cobre en los hepatocitos debida a la reducción de su excreción biliar. La consecuente tesaurismosis alcanza proporciones tan elevadas como 20 veces la normal. Después sobrevienen las alteraciones histológicas principales: necrosis hepatocelular, inflamación portal y periportal, y fibrosis. Puede acaecer la necrosis masiva con insuficiencia hepatocelular fulminante. En otros casos la hepatopatía es apenas leve. La liberación masiva del cobre produce hemólisis; la movilización lenta del cobre hepático conduce a su depósito en otros órganos, primariamente el sistema nervioso central, las córneas y los riñones. En esta etapa de la historia natural de la enfermedad la cantidad de cobre en el hígado disminuye a niveles normales (l). No hay signos clínicos específicos o diagnósticos. Uno de los rasgos de esta enfermedad es que no hay dos pacientes iguales aún dentro de una misma familia. Actualmente se hace el diagnóstico más temprano y, por consiguiente, se ve menos los anillos de Kayser-Fleischer o los signos neurológicos graves. Dentro de la variedad de manifestaciones clínicas predominan las hepatopatías y los trastornos neuropsiquiátricos.
La mayoría de los pacientes tienen algún grado de hepatopatía que se revela entre los 8 y l8 años de edad, aunque la cirrosis puede estar presente en niños menores de 5, o hacerse sintomática recién sobre los 60 años de edad. Las formas clínicas agudas son indiferenciables de las debidas a otras causas de hepatopatias agudas( hepatitis agudas, hepatitis aguda fulminante, hepatitis aguda asociada a hemólisis). La biopsia hepática, especialmente coloreada con Rhodanina, puede demostrar los depósitos focales de cobre en la minoría de los pacientes.
Son notorios por su ausencia los anillos de Kayser-Fleischer, las anomalías neurológicas y los niveles bajos de ceruloplasmina sérica. Los anillos mencionados faltan en 50% de los pacientes con la modalidad clínica de hepatitis crónica; para aumentar las dificultades es pertinente mencionar que ellos pueden estar presentes en otras hepatopatías) cirrosis biliar primaria, colangilitis esclerosante primaria, hepatitis autoinmunes y determinados síndromes colestásicos familiares).
Los síndromes neurológicos comienzan en la adolescencia y en la segunda década de la vida, pero pueden retrasarse hasta la cuarta década. Los anillos de Kayser-Fleischer facilitan el diagnóstico en grado tal que es suficiente la medición del nivel de ceruloplasmina sérica para establecer el diagnóstico definitivo.
Las anormalidades psiquiátricas pueden ser las únicas expresiones clínicas en un 20%, o las primeras en aparecer en un 30% (2,3).
El avance tecnológico a nivel de moléculas e imágenes está demostrando la necesidad de revisar los parámetros clínicos y de laboratorio tradicionalmente empleados para el diagnóstico. No obstante que la causa sea la disfunción de una enzima, la relación entre fenotipo y genotipo puede ser tan complicada que debemos seguir vigilando la presentación clínica en formas desacostumbradas (4).
Se ha identificado más de 40 mutaciones del gen ATP7B, responsable de la enfermedad. La mayoría de los pacientes lleva dos mutaciones diferentes en cada uno de sus dos cromosomas. Se explica, consecuentemente, que la tecnología actual no haya desarrollado un método de laboratorio sencillo y reproducible en todos los casos. Sigue siendo imprescindible examinar cuidadosamente a los pacientes buscando los anillos corneales, los signos neurológicos típicos, la disminución de ceruloplasmina sérica, el aumento de la excreción urinaria de cobre y la elevación del contenido cúprico en el hígado, reconociendo, sin embargo, las variaciones dependientes de la historia natural de la enfermedad de Wilson tanto como las mutaciones en cada individuo. Las dificultades son mayores para el diagnóstico de las modalidades hepatológicas; empero, en la forma neurológica pueden faltar los anillos de Kayser-Fleischer hasta en l0% y resultar normales los niveles de ceruloplasmina sérica en l5%. En esta era de avances tecnológicos sorprendentes tienen que mover a reflexión las muy recientes citas textuales que siguen. " El examen clínico es más sensitivo que cualquier otro método diagnóstico" (2). " En ausencia de signos clínicos típicos ningún método de laboratorio aislado permite el diagnóstico certero de enfermedad de Wilson" (1). " En el momento actual lo primero es plantear el diagnóstico y lo siguiente usar e interpretar apropiadamente los métodos de investigación clínica que han soportado la prueba del tiempo " (5).
OBSERVACIÓN CLÍNICA
ANAMNESIS:
Mujer, de 26 años de edad, nacida y residente en el Callao, soltera y sin embarazos. Profesora de educación inicial. Sus padres y dos hermanos se encuentran sanos. En abril de 1998, los familiares la notaron desinteresada en su cuidado personal, con tendencia a permanecer callada y no expresar sus estados afectivos, reacia para visitar a sus familiares y amistades. Disminuyó considerablemente su colaboración en las actividades diarias del hogar. No hubo modificaciones en el apetito ni en el ritmo del sueño. Estos síntomas se desarrollaron en el lapso de dos meses. Después aparecieron movimientos rápidos en la mano derecha, a manera de sacudidas, especialmente al realizar movimientos intencionales tales como manipular cubiertos, escribir, usar tijeras y emplear la escoba. Estas molestias se intensificaron progresivamente interfiriendo en grado alto los actos más elementales; la escritura se volvió micrográfica e incoordinada groseramente. Las dificultades en la marcha evolucionaron insidiosamente, caracterizándose por rigidez al caminar, giros lentos en cada uno de los pasos con rotación externa de las caderas y rotación interna de las piernas, lo que configuraba una apariencia " saltarina " al caminar. Un médico interpretó sus trastornos como psicogénicos, prescribiéndole ansiolíticos y antidepresivos durante dos meses, sin obtener mejoría. A los 6 meses del comienzo clínico de la enfermedad se encontraba disártrica, con acentuada incoordinación de los miembros, había sialorrea y disfagia cervical. Su expresión facial era burlona o irónica. Las sacudidas en la mano derecha eran grotescas y consistían en movimientos rápidos a manera de un aleteo cuando levantaba el brazo o trataba de aprehender objetos, que terminaban cayendo por la dismetría y torpeza extremas. En otro centro hospitalario se planteó el diagnóstico diferencial con parkinsonismo juvenil y temblor esencial, un electroencefalograma y una tomografía axial computarizada del cerebro fueron interpretados como normales y le prescribieron L-Dopa y carbidopa (125 mg cada 6 horas) y propanolol (40 mg cada 8 horas) durante dos meses sin modificación positiva de los signos. Al año de enfermedad, uno de nosotros, tuvo la oportunidad de entrevistarla, y con los resultados de la investigación y los hallazgos neurológicos solicitó estudios del cerebro con resonancia magnética y dosaje bioquímico de ceruloplasmina y de cobre en suero, así como excreción de cobre en orina de 24 horas. Infortunadamente, la indigencia económica de la familia imposibilitó la obtención de los exámenes solicitados. No se supo de la paciente hasta 10 meses después. Esta vez fue traída por su padre en brazos y no podía mantenerse de pie. Su expresión facial, con la boca entreabierta, correspondía a una sonrisa burlona. Su mutismo impedía evaluar el estado mental. La disfagia se había agravado y producido un grave estado de desnutrición. En estas condiciones fue hospitalizada.
HALLAZGOS DEL EXAMEN FÍSICO
18.03.2000. Luce muy adelgazada y con considerable atrofia de las masas musculares. Frecuencia cardiaca: 100/min; Frecuencia respiratoria: 20/min; Presión arterial: 110/70 mm Hg y Temperatura: 36.8°C. No se pudo determinar el peso ni la talla, debido a la gravedad y discapacidades motoras de la enferma. En los ojos era evidente un anillo de color marrón intenso a nivel del limbo esclerocorneal, con mayor notoriedad en el polo superior. La exploración neurológica reveló todo lo que sigue: las pupilas de un tamaño de 9mm, isocóricas y normoreactivas. Oftalmoparesia marcada, tanto a la mirada conjugada horizontal como en dirección vertical, y ausencia de la convergencia ocular. El fondo del ojo era normal. Diplejía facial central, hiporreflexia corneal, la fuerza muscular de los maseteros normales, aunque lentos y dispráxicos. Paresia bilateral del velo del paladar y el reflejo nauseoso disminuido. Distonía de la lengua durante la protrusión; sin atrofia ni fasciculaciones. Rigidez del cuello en todas las direcciones, durante el movimiento pasivo. Cuadriparesia con marcada rigidez plástica. Hiporreflexia osteotendinosa generalizada. No se encontró signos de Babinski ni de Hoffmann. Reflejos cutáneo-abdominales presentes. Temblor intencional en los miembros superiores, con marcada dismetría y adiadococinesia.
Con la maniobra de Barré, los miembros presentaban temblor, con fenómeno de aleteo grosero en la mano derecha, que desaparecía en condición de reposo. Postura distónica del cuello en láterocolis. Distonía a nivel de las muñecas y actitud en flexión en las articulaciones metacarpofalángicas. Las piernas se encontraban espontáneamente hiperextendidas en extremo, adoptando una postura en tijeras, con rotación interna e hiperpronación a nivel de los tobillos. Era permanente la expresión de sonrisa irónica. Con una frecuencia imposible de precisar, tenia accesos de oculogiria derecha y supraversión ocular, durante l0 a l5 segundos, con remisión espontánea (Figuras 1 y 2). No se pudo explorar adecuadamente la sensibilidad, debido a las condiciones generales de la paciente.
La exploración por sistemas fue semiológicamente normal.
RESULTADOS DE EXÁMENES AUXILIARES
Sangre:
Hematíes 3'660,000/ul; Leucocitos 3,100/ul; Hb: 10.6 g/dl; Ht: 30.4%.
Eosinófilos:1%; Basófilos: 2%; Abastonados 4%;Linfocitos: 13%.;
Monocitos 2% Neutrófilos :68%. Glucosa: 106 mg%; Urea: 17 mg/dl;
Creatinina: 0.81 mg/dl Orina:. reacción ácida; glucosa, proteínas, bilis y cuerpos cetónicos: negativo. Bioquímica sanguínea:
Tiempo de tromboplastina parcial: 35 seg. Tiempo de protrombina : 10.8
seg. Fibrinógeno: 204.5 mg/dl.Tiempo de sangría:3 min.30 seg. Antígeno
de superficie para el virus de la hepatitis B: no reactivo. Proteínas
totales: 7.10 g/dl. Albúmina: 3.5 g/dl; Globulinas: 3.6 g/dl. Cobre
sérico: 72.2 ug/dl. (70-150 ug/dl). Ceruloplasmina. 6.1 mg/dl (10-35
mg/dl). Cobre en orina de 24 horas: 77.7 ug/L ( 0-60 ug/L).
Ecografía abdominal:
hígado de tamaño normal, superficie y borde romo; sistema bilioportal
de caracteres normales. El parénquima hepático de ecodensidad
heterogénea difusa.Otros órganos normales. Conclusión: hepatopatía
difusa. Laparoscopía: El lóbulo derecho del hígado de
superficie irregular, nodular, de color vinoso pálido y de bordes romos.
El lóbulo izquierdo con moderado incremento del tamaño, de similares
características al derecho, aunque el borde no se encuentra romo. El
bazo y las vías biliares de aspecto normal. No ascitis. Diagnóstico
Laparoscópico: Cirrosis hepática micronodular. Biopsia hepática: cirrosis hepática. Resonancia Magnética del Encéfalo:
Signos de atrofia frontal y temporal bilateral con predominio
izquierdo, señales de hiperintensidad en cortes axiales y coronales en
T2, que comprometen los núcleos de los ganglios basales, ambos tálamos,
el tronco cerebral, los pedúnculos cerebrales y los núcleos dentados del
cerebelo (Figuras 4,5,6).
Consulta oftalmológica: evidencia del anillo de Kayser-Fleischer en la membrana de Descemet, mediante la lámpara de hendidura (Figura 3).
Consulta gastroenterológica:
evidencia de membrana cricofaríngea con lumen infranqueable para una
bujía de calibre l9 Fr; el tratamiento con dilatadores Savary permitió
pasar hasta el calibre 4l Fr.
|
Figura 1. Crisis de láterocolis con oculogiria derecha y supraversión ocular. |

|
Figura 2. Expresión facial típica en forma de sonrisa irónica. Posición distónica a predominio distal en los miembros y postura rígida en las piernas en forma de tijeras. |
|
Figura 3. Presencia del anillo de Kayser-Freischer en la membrana de Descemet, de color marrón claro. |

|
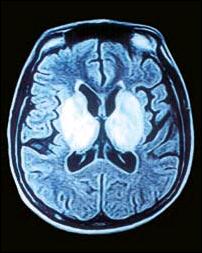
Figura 4. Señales de hipertensidad en T2 en la RM que muestra atrofia fronto temporal assimétrica y considerable compromiso de los ganglios basales y los tálamos. |
|
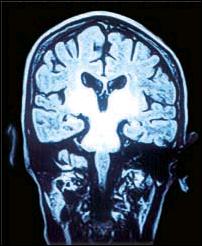
Figura 5. IRM en corte coronal (T2) donde se muestra la extensión del proceso degenerativo hacía niveles interiores del tronco encefálico. |
|
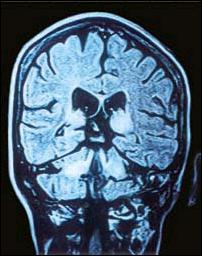
Figura 6. Corte coronal posterior (T2) que evidencia afectación de los núcleos dentados del cerebelo, los pedúnculos cerebelosos superiores y los tálamos. |
EVOLUCIÓN CLÍNICA Y TRATAMIENTO
Durante su hospitalización (Hospital Nacional "Dos de Mayo"), recibió por sonda nasogástrica el régimen alimenticio adecuado para enfermedad de Wilson; se utilizó antibióticos para controlar infecciones intrahospitalarias.Después de confirmar el diagnóstico de Enfermedad de Wilson, se administro D-penicilamina, inicialmente 500 mg/día, hasta llegar a una dosis de mantenimiento de 1,500/dia/oral. A los 45 días de su internamiento, fue dada de alta, en mejores condiciones generales y transferida al Instituto Nacional de Rehabilitación. Una reevaluación reciente (Febrero 200l), por uno de los autores (MAC), permitió constatar: un mejor estado mental, al obtener 26 puntos en el test minicognitivo de Folstein. Continúa disártrica. Presentó disfagia hasta que fue sometida a procedimiento para corrección de la estenosis esofágica, con lo cual pudo ingerir sus alimentos en condiciones normales. No existen defectos del lenguaje comprensivo ni expresivo. Las pupilas persisten midriáticas, con un tamaño de 9 mm, isocóricas e hiporeactivas; mejoría de la oftalmoparesia y actualmente se observa nistagmo horizontal y vertical. No ha vuelto a presentar crisis oculogira con supraversión. Aún existe cuadriparesia; sin embargo, la rigidez del cuello y de los miembros ha mejorado. El fenómeno de aleteo de la mano derecha aún persiste. La rigidez extrapiramidal y los fenómenos distónicos son de menor intensidad. No existen signos de piramidalismo y la exploración de la sensibilidad en todas sus formas es normal. El temblor intencional, la dismetría y la adiadococinesia persisten. El fondo del ojo es normal y no existe alteración en el control de los esfínteres.
DISCUSIÓN Y COMENTARIOS
Los síntomas psíquicos, los signos neurológicos, la presencia del anillo de Kayser - Fleischer, las cifras disminuidas de la ceruloplasmina sérica, la excreción aumentada del cobre en la orina de 24 horas y la presencia de cirrosis hepática subclínica nos han permitido hacer el diagnóstico de enfermedad de Wilson que motiva la presente comunicación. Es importante resaltar, en el inicio de la enfermedad, las manifestaciones psíquicas; la abulia, el desinterés para la comunicación, la pobreza de las respuestas afectivas, el descuido en su aseo personal, y la propensión al mutismo; todas ellas indicando con claridad, trastornos funcionales o lesionales de las regiones frontales anteriores. Las alternativas para el diagnóstico diferencial inicial, incluyen probable consumo de sustancias tóxicas; considerando la edad de la paciente; el desarrollo de un proceso expansivo con esta localización, el inicio de alguna variante clínica de la leucoencefalopatía esclerosante subaguda de Van Bogaert, y, finalmente, un desorden afectivo depresivo, o sintomatología atribuible a psicosis esquizofrénica. De allí que el planteamiento inicial de estos síntomas como atribuibles a enfermedad de Wilson se hace difícil, aún para clínicos muy experimentados. La aparición del temblor asimétrico, intencional, grosero y con aleteo en la mano derecha, debe orientar a la sospecha de degeneración hepato-lenticular, especialmente, cuando no se constata nistagmo, palabra escandida o disdiadococinesia inicial, sugerentes de afecciones cerebelosas. El temblor de reposo, característico del parkinsonismo, puede estar presente ocasionalmente en la enfermedad de Wilson; sin embargo, se insiste en la presencia del temblor asimétrico, burdo y con aspecto de aleteo, como característico en esta enfermedad. (LeWitt y Brewer, 1994 ). Las razones por las que algunos pacientes, desarrollan parkinsonismo con fenómenos distónicos mientras que otros son más afectados por disfunción bulbar y temblor no han sido explicados (Denny-Brown, 1964).
La enfermedad de Wilson está vinculada a una alteración progresiva de las funciones cognitivas, mediante un proceso de desaferentación de los lóbulos frontales, como consecuencia de la lesión de los ganglios basales (6), fenómeno denominado diaschisis; así es posible explicar las manifestaciones iniciales neuropsiquiátricas de la enfermedad en esta paciente, al revelar alteraciones conductuales de la motivación y del estado afectivo.
La resonancia magnética del cerebro (RM) puede ser útil para demostrar la gravedad de las alteraciones del sistema nervioso central. Las anomalías más comunes son cambios en la intensidad de las señales de las substancias gris y blanca, así como atrofia del núcleo caudado,pedúnculo, hemisferios cerebrales y cerebelosos (7). También se han descrito señales de hipointensidad en T1 e hiperintensas en T2, afectando los ganglios basales y otras estructuras adicionales en relación con la severidad y duración del cuadro clínico (8,9).
Este procedimiento empleado en nuestra paciente nos permitió constatar la presencia de atrofias en las regiones frontotemporales a predominio izquierdo, compromiso degenerativo (señales hiperintensas potenciadas en T2) de los ganglios basales, los tálamos, el tronco cerebral, los núcleos dentados del cerebelo y los pedúnculos cerebelosos en su sinapsis con las regiones talámicas. Sobre esta base parece posible interpretar las manifestaciones neurológicas. Así, el parkinsonismo y los fenómenos distónicos de esta paciente serían explicables por el compromiso lentículo-estriado y de las eferencias pálido-putaminales al tálamo y al córtex motor extrapiramidal (Figuras 4 y 5). La combinación del temblor y la dismetría puede estar relacionada con el daño de las conexiones de los núcleos dentados del cerebelo y los núcleos ventrolaterales del tálamo a través de los pedúnculos cerebelosos superiores (Figura 6 ).En otras observaciones existe correlación del pseudoparkinsonismo con las anormalidades del estriado o del tracto pontocerebeloso, y de los signos cerebelosos con las alteraciones del tracto dentadotalámico. La presencia de derivaciones portosistémicas se asocia notablemente con alteraciones del globo pálido (10) Los accesos distónicos cefálicos con supraversión ocular derecha (Figura 2), han sido observados por Segawa y sus colaboradores (11,12) en la entidad denominada distonía progresiva hereditaria, probablemente autosómica dominante, asociada a parkinsonismo, generalmente presente en infantes. La interpretación fisiopatológica de las pupilas midriáticas, la oftalmoparesia y los defectos en la convergencia en nuestra paciente puede sustentarse en la perturbación del tronco encefálico de los núcleos de Edinger Westphal, los correspondientes a los óculomotores y los de Perlia, implicando también, los fascículos de conexión de las estructuras antes mencionadas. La cuadriparesia con rigidez plástica y las posturas distónicas de los 4 miembros pueden ser la expresión del compromiso córtico-subcortical y de los de la corteza motora extrapiramidal, así como, de las eferencia putaminales al tálamo. Finalmente el desarrollo progresivo del mutismo por anartria bulbar y la distonía lingual se explicarían por el compromiso de grupos nucleares ubicados en los ganglios basales, los tálamos y los núcleos rojos, encargados del control regulador de los movimientos involuntarios y el tono muscular de las estructuras bulbares, a través del fascículo central de la calota, encargado de la eferencia de los impulsos neurales de las estructuras antes mencionadas. La ausencia de atrofia y fasciculaciones en la lengua descarta lesión primaria en los núcleos bulbares.
La exploración oftalmológica con la lámpara de hendidura demostró en la membrana de Descemet, la presencia del anillo de Kayser- Fleischer (Figura 3), signo cardinal para el diagnóstico de la forma neurológica de la enfermedad de Wilson.
El depósito excesivo de cobre en esta enfermedad, se debe a que el cobre intrahepatocitario no accede a la circulación sistémica por déficit de la ATPasa, que lo incorpora a la ceruloplasmina (13); existe disminución en su excreción biliar, aunque su absorción intestinal y su transporte son normales. Estas alteraciones se deben a la mutación del gen ATP7B, relacionado también con la enfermedad de Menkes (otra rara enfermedad del metabolismo del cobre). Las bajas concentraciones de ceruloplasmina que se asocian a la EW son consecuencia de la alteración en su síntesis y no tienen ninguna relación patogénica directa con la enfermedad (14). Esta observación y afirmación se confirma considerando que el gen de la ceruloplasmina se localiza en el cromosoma 3 y el gen ATP7B en el cromosoma l3 (15). La biopsia hepática permitió comprobar la presencia de cirrosis hepática, considerándose las observaciones histopatológicas en este caso como no patognomónicas en fases iniciales(16); con el microscopio óptico, puede detectarse esteatosis macrovesicular, depósitos de hemosiderina, acumulación de glucógeno intranuclear e hipertrofia de las celulas de Kupffer; en estadíos más evolucionados, aparece necrosis hepatocitaria con colapso reticular y finalmente cirrosis macronodular. Se ha descrito alteraciones características en las mitocondrias con el microscopio electrónico(17) sugiriendo la formación de radicales libres y el daño oxidativo, por defecto mitocondrial que expliquen la patogénesis de la acumulación del cobre en la enfermedad de Wilson. En la presente observación se recomienda, la búsqueda de los portadores asintomáticos en los miembros de la familia más cercanos, mediante el screening del metabolismo del cobre o los estudios genéticos orientados a la búsqueda de las mutaciones en los genes mencionados anteriormente en los cromosomas 3 y 13 respectivamente.
CONCLUSIONES
La presente observación clínica reúne suficientes criterios para sustentar el diagnóstico de enfermedad de Wilson con predominio de las manifestaciones neuropsiquiátricas, sobre las de la cirrosis hepática subclínica. El estudio del encéfalo con resonancia magnética nos ha permitido, por una parte, encontrar una relación clínica directa de la gravedad del caso con los hallazgos de este procedimiento (RM) y, por otra parte, plantear los probables mecanismos fisiopatológicos de los trastornos neurológicos y psíquicos de la enfermedad. Sugerimos considerar el diagnóstico de enfermedad de Wilson (EW) desde un inicio en un paciente joven con manifestaciones conductuales, acompañados o seguidos de temblor asimétrico intencional y en aleteo, asociados a parkinsonismo. El tratamiento utilizando D-penicilamina, trientina o sales de Zinc indefinidamente permite modificar favorablemente la historia natural de la EW. El diagnóstico temprano, especialmente de los homozogitos asintomáticos, es fundamental para evitar la progresión a lesiones estructurales irreversibles.
La asociación con membrana cricofaríngea causante de estenosis cervical intensa no ha sido comunicada con anterioridad.
BIBLIOGRAFÍA
1. STEINDL, FERENZI P, DIENES HP et al. Wilson's disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology 1997; 113:212-218.
2. FERENCI P. Wilson's disease. Clin Liver Dis l998; 2: 31-49.
3. STERNLIEB I. Wilson's disease. Clin Liver Dis 2000; 4:229-239.
4. DUFOUR J-F, KAPLAN MM. Muddying the water: Wilson's disease challenges will not soon disappear. Editorial. Gastroenterology 1997; 113: 348-349.
5. SCHILSKY ML, STERNLIEB I. Overcoming obstacles to the diagnosis of Wilson's disease. Editorial. Gastroenterology 1997; 113: 350-353.
6. FEENEY DM,BARON JC. Diaschisis.Stroke 1986; 17.817-830.
7. VAN WASSENAER-VAN HALL HN, VAN DER HEUVEL AG, ALGRA A,et al. Wilson's disease: findings at MR imaging and CT of the brain with clinical correlation. Radiology 1996; 198 :531-536.
8. NAZER H, BRISMAR J, AL-KAWI MZ, GUNAZEKARAN TS. Magnetic Resonance imaging of the brain in Wilson's disease. Neuroradiology 1993; 35:134-141.
9. THUOMAS KA, AQUILONIUS, BERGSTROM K, WERSTERMARK K. Magnetic resonance imaging of the brain in Wilson's disease.Neuroradiology 1993; 35: 134-141.
10. GRIMM G, MADL CH, KATZENSCHLAGEN E, et al. Detailed evaluation of brain dysfunction in patients with Wilson's disease. EEG Clin Neurophysiol 1992; 82: 119-124.
11. SEGAWA M, HOSAKA A, MIYAGAWA et al.Hereditary progressive distonia with marked diurnal fluctuation. Adv Neurol 1976; 14: 235-245.
12. SEGAWA M, NOMURA Y, TANAMA S et al.Hereditary progressive dystonia with marked diurnal fluctuation: Consideration on its pathophysiology based on the characteristics of clinical and polysomnographical findings. Adv. Neurol 1998; 50: 367.
13. SCHILSKY ML. Wilson's disease: Genetic basis of copper toxicity and natural history. Semin Liver Dis 1992; 16: 83-95.
14. ZUCKER SD, GOLLAN JL. Wilson's disease and hepatic copper toxicosis. En: Zakim D, Boyer TD, editors. Hepatology: a textbook of liver disease. Filadelfia: WB Saunders,1996; 1405 - 1439.
15. FRYDMAN M, BONNE-TAMIR, FARRER LA et al. Assignment of the gene of Wilson's disease to chromosome 13: linkage to the esterase D locus. Proc Natl Acad Sci USA 1985; 82 : 1819-1821.
16. CUZA E, MAIER DOBERSBERGER, POLLI C, et al. Screening for Wilson's disease by serum ceruloplasmin. J Hepatology 1997; 27: 358-362.
17. GU M, COOPER JM, BLUTER P et al. Oxidative-phosphorylation defects in liver of patients with Wilson's disease. Lancet 2000; 356: 469-470.