REPORTE DE CASO CLÍNICO
Hepatitis granulomatosa tuberculosa como causa de fiebre de origen desconocido
David Loja Oropeza1; Maricela Vilca Vásquez2; Pierino Alvarez Bedoya3.
1 Departamento de Medicina Interna. Hospital Arzobispo Loayza. Lima-Perú.
2 ESSALUD
3 Departamento de Patología. Hospital Arzobispo Loayza. Lima-Perú.
RESUMEN
Presentamos el caso clínico de una paciente con fiebre de origen desconocido por hepatitis granulomatosa de etiología tuberculosa.
Se trata de una paciente de 50 años de edad con 50 dias de enfermedad caracterizada por escalofríos, fiebre de 39 °C y diaforesis profusa. Antecedente de malaria en siete ocasiones. Al examen de ingreso se encontró una paciente adelgazada y pálida. En la evolución desarrolló pancitopenia, hepatoesplenomegalia masiva, ictericia y anasarca. Se hicieron pruebas de tamizaje para infecciones, neoplasias, colagenosis y enfermedades granulomatosas. Los exámenes de laboratorio mostraron disociación fosfatasa alcalina-transaminasas y fue la pista que condujo al diagnóstico final de tuberculosis mediante el estudio histológico del parénquima hepático. El tratamiento específico para tuberculosis produjo remisión de la fiebre, ascitis y hepatomegalia, y normalización de las pruebas hepáticas con evolución clínica satisfactoria.
Palabras clave: 1. Fiebre de origen desconocido 2. Tuberculosis hepática 3. Granulomatosis hepática 4. Colestasis intrahepática.
SUMARY
The clinical case of one patient with fever of unknown origin, due to granulomatous hepatitis of tuberculous etiology was presented.
The patient was a a 50-year-old woman, with 50 days illness characterized by chills, 39°C fever and heavy diaphoresis. She had a record of seven malaria cases. She looked thin and pale at the initial physical examination. During the evolution, she developed pancytopenia, massive hepatosplenomegaly, jaundice, and anasarca. The patient underwent screening tests for infection, neoplasias, collagenosis, and granulomatous diseases. The laboratory tests showed transaminase-alkaline phosphatase dissociation, which led to the final diagnosis of tuberculosis, through the histological examination of the liver parenchyma. The specific treatment against tuberculosis caused remission of fever, ascites, and hepatomegaly and normalization of liver tests, with satisfactory clinical evolution.
Key Words: Fever of unknown origin, hepatic tuberculosis, hepatic granulomatosis, intrahepatic cholestasis.
INTRODUCCIÓN
Uno de los mayores desafíos para el clínico lo constituye la fiebre de origen desconocido (FOD) para cuyo discernimiento no existen algoritmos ni pistas diagnósticas fiables (1). El enfoque se dificulta cuando las manifestaciones clínicas son mínimas, sutiles o insuficientes para configurar una enfermedad o un grupo de enfermedades.
Clásicamente se define FOD como aquella enfermedad de más de tres semanas de duración con temperatura que supera los 38. 3°C y cuyo diagnóstico permanezca incierto, luego de una semana de estudios (1-3).
En las series de las dos últimas décadas se han reportado como 200 entidades clínicas causantes de FOD, de ellos el 70% corresponden a procesos infecciosos, neoplasias o colagenosis; siendo las condiciones más mencionadas: tuberculosis, endocarditis, linfoma, tumores sólidos, enfermedad de Still del adulto, vasculitis, lupus eritematoso sistémico y enfermedades granulomatosas (2-6).
A propósito del presente caso de FOD, ocasionado por una hepatitis granulomatosa de origen tuberculoso, se realiza una revisión de la literatura sobre esta entidad con comentarios sobre enfoques de diagnóstico y tratamiento.
CASO CLINICO
Paciente mujer de 50 años de edad, natural de Bolivar, La Libertad, procedente de Lima, Testigo de Jehová, admitida en el Hospital Arzobispo Loayza en junio de 1996 con 50 dias de enfermedad caracterizado por fiebre de 39°C, precedida de escalofríos y seguida de diaforesis profusa. Un mes antes del ingreso había notado máculas hipercrómicas muy pruriginosas, no confluentes, de 2 mm de diámetro, en todo el cuerpo, que mejoró con sintomáticos. Una semana después le diagnostican fiebre tifoidea con aglutinaciones en el límite superior recibiendo un curso de cotrimoxazol, sin mejoría. Fue investigada para tuberculosis pulmonar y malaria, resultando negativa. Recibió cursos con amoxicilina y luego ampicilina ante la presunción de infección respiratoria alta. Al persistir la fiebre acude al Hospital donde es internada.
Funciones biológicas: Orinas oscuras. Sueño alterado por la fiebre. No variación ponderal.
Antecedentes: Salpingo oforectomía bilateral 15 años atrás. Durante su niñez padeció de paludismo en siete oportunidades con tratamientos incompletos. A los 40 años de edad fue diagnosticada de anemia. Un año después tuvo un nódulo tiroideo sin evidencia de malignidad en su histología.
Examen físico: Febril, crónicamente enferma, adelgazada y pálida. Se aprecian cicatrices queloideas en la cara anterior del cuello y en la línea media infraumbilical. Pápulas hipercrómicas en piernas y pies. El abdomen dejaba palpar el bazo a 5 cm debajo del reborde costal, de superficie lisa, bordes regulares y de consistencia normal. El resto sin alteraciones.
Evolución
Desde el cuarto día de hospitalización se incrementa el tamaño del bazo hasta alcanzar 10 cm debajo del reborde costal. En el intercurso se documenta infección de vías urinarias causada por E. Coli que fue tratada satisfactoriamente con ciprofloxacino, sin embargo continúa con fiebre y desarrolla pancitopenia. Gota gruesa en varias ocasiones y test de Marañón negativas. El mielograma fue informado como hiperplasia reticular leve. La serología para HIV, fiebre tifoidea, brucellosis, mononucleosis y toxoplasmosis fueron negativos. Las baciloscopías para tuberculosis, los hemocultivos y el mielocultivo no fueron contributorios. Una ecografía abdominal mostró esplenomegalia moderada y formaciones líquidas pequeñas en cabeza y cuerpo de páncreas. Se obtuvo líquido ascítico tipo trasudado con adenosin deaminasa y Papanicolaou negativos. La bioquímica hepática reveló hiperbilirrubinemia a predominio directo con disociación fosfatasa alcalina-transaminasas incipiente.
En la segunda semana de hospitalización la paciente se torna ictérica, desarrolla anasarca y el hígado se palpa a 14 cm debajo del reborde costal. Una TAC abdominal evidenció hepatoesplenomegalia importante sin colecciones intraabdominales. También fueron negativos las pruebas de alfa-fetoproteina, marcadores para hepatitis viral, citomegalovirus y lues.
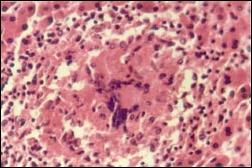
Fue diagnosticada de absceso periodontal y ascaridiasis mejorando con el tratamiento. Una nueva ecografía abdominal demostró hepatoesplenomegalia, ascitis, vena porta dilatada y en el perfil hepático de control se encontró inversión de la relación albúmina-globulina y una acentuada disociación fosfatasa alcalina-transaminasas e hiperbilirrubinemia indirecta. La endoscopía alta reportó várices esofágicas grado II. Se procedió a la biopsia hepática por laparoscopía. El cultivo de tejido hepático fue negativo para Mycobacterium tuberculoso y su histología resultó compatible con hepatitis granulomatosa de origen tuberculoso (Fig. 1 y 2). Se inició el tratamiento para tuberculosis, la fiebre cayó al octavo día y las pruebas hepáticas se normalizaron al segundo mes de tratamiento con remisión de la ascitis y hepatomegalia. Seis años después el bazo no ha disminuido de tamaño. Hay evidencia de hipertensión portal, persistencia de várices esofágicas y ha desarrollado plaquetopenia.
Figura 1

Figura 2
DISCUSIÓN
Recientemente Durack y Street debido al incremento de pacientes con FOD asociada a métodos invasivos, a neutropenia o a infección por VIH propusieron una nueva clasificación que incluye FOD Clásica, FOD Nosocomial, FOD Neutropénica y FOD asociada al VIH (3,7). Otras categorías a considerarse son FOD en Cáncer, FOD en ancianos, FOD en niños. Ello permite elaborar un diagnóstico diferencial dirigido a cada grupo.
El caso en discusión corresponde al grupo de FOD Clásica. En este grupo algunas consideraciones útiles a tenerse en cuenta para el diagnóstico serían: El patrón o la duración de la fiebre no se relacionan con el diagnóstico final como por ejemplo no son frecuentes como se espera el patrón característico de malaria, la neutropenia cíclica o la fiebre de Pel-Ebstein del linfoma. Igualmente la disociación pulso-temperatura que suele verse en fiebre tifoidea, brucellosis, enfermedad por Legionella, fiebre por drogas, linfoma, fiebre facticia, por lo general está ausente y solo es útil cuando está presente. Se sospechará de la presencia de tumores sólidos o de enfermedad de Still cuando la prueba del naproxeno resulte positiva. La triada de fiebre alta, erupción evanescente y artritis en un adulto joven con ferritina sérica elevada en 10 veces es también indicadora de enfermedad de Still. Otra pista útil en caso de linfomas lo constituye un marcado incremento de la b2-microglobulina o de la deshidrogenasa láctica séricas por encima de 1000 UI asociada a la presencia de adenomegalias, esplenomegalia y anemia o trombocitopenia. En ancianos con síntomas sistémicos, fiebre y eritrosedimentación elevada considerar la posibilidad de biopsiar la arteria temporal incluso en ausencia de signos específicos de arteritis (5,8).
De acuerdo al diagnóstico final las etiologías del FOD pueden agruparse en cinco categorías: 1. Infecciosas. 2. Neoplasias. 3. Colagenosis. 4. Enfermedades granulomatosas. 5. Misceláneas. Un tercio de las FOD correlacionan con infecciones siendo las más frecuentes tuberculosis, endocarditis subagudas, infecciones intraabdominales, virosis y micosis sistémicas. De ellas la infección más común es la tuberculosis, constituyendo el 40% de las infecciones en las series españolas(8)
El enfoque diagnóstico de las FOD debería ser secuencial:
1. Historia clínica y examen físico minucioso que permita una presunción diagnóstica y de no ser contributorio disponer del hemograma, velocidad de sedimentación globular, examen completo de orina, pruebas de función hepática, proteinograma electroforético, radiografía de tórax y abdomen, PPD, serología para salmonella, brucella y VIH, cultivos (orina, sangre, heces), si es mujer anticuerpos antinucleares, ecografía abdominal y rastreo corporal con galio-67 o indio-111 si amerita el caso.
2. Si aún no se tiene orientación diagnóstica hospitalizar al paciente para exploración física diaria, buscando la aparición de algún nuevo signo y descarte de fiebre facticia y por drogas.
3. Realizar punción lumbar, biopsias (médula ósea, hígado, arteria temporal, piel, músculo) arteriografía, laparoscopía y otras como ecocardiograma, TAC cerebral. 4. Si no se llega a una orientación diagnóstica quedan dos posibilidades. Si el estado general es bueno y no hay pérdida ponderal se recomienda dar de alta y reevaluar en 3 y 6 meses. Si el estado general es malo o existe deterioro progresivo evaluar la realización de una laparatomía exploratoria, si cursa con síntomas abdominales persistentes, masa abdominal palpable, ascitis de causa no determinada, radiografía o ecografía patológicas, hepato y/o esplenomegalia, derrame pleural, aumento de fosfatasa alcalina o pérdida de peso mayor del 10%.
Entre algunas actitudes terapéuticas a pesar de no tener un diagnóstico de certeza, son el uso de tuberculostáticos como prueba terapéutica durante seis semanas, la fiebre suele caer en la primera semana. Asimismo, el uso de antibióticos a dosis altas por la posibilidad de endocarditis con cultivos negativos;corticoides en caso de polimialgia reumática con biopsia de arteria temporal normal, si hay respuesta en horas es casi diagnóstica; ácido acetilsalicílico en caso de fiebre reumática o enfermedad de Still, aunque en ésta suele ser necesario administrar corticoides para que caiga la fiebre; y cloroquina ante la sospecha de absceso hepático amebiano (5,8)
A pesar de los procedimientos diagnósticos un 5-10% quedan como auténticos FOD con buena evolución y resolución espontánea de la fiebre en la mayoría de los casos. De este grupo solo un 10% tendrá un diagnóstico definitivo en el seguimiento a 5 años y la mortalidad alcanza solo un 3% (5,8,9)
Como podemos apreciar nuestra paciente no presentaba cuadro clínico característico de alguna enfermedad y precisamente las FOD son eso, enfermedades comunes pero de presentación atípica. Secuencialmente el estudio del caso incluyó hasta el tercer estadío.
Cuando tenemos un paciente con fiebre, ictericia y hepatoesplenomegalia, algunos planteamientos diagnósticos a tomarse en cuenta incluyen SIDA, lupus eritematoso sistémico, artritis reumatoidea, sarcoidosis, malaria, tuberculosis, mononucleosis, hepatitis citomegálica, leucemias mielocíticas y linfocíticas crónicas entre otros, que fueron posibilidades que se alejaron de acuerdo al cuadro clínico y los exámenes solicitados, quedando como prioridades enfermedad tuberculosa y neoplasia hematológica.
En relación a la enfermedad tuberculosa, el espectro clínico es variado, pudiendo afectar a cualquier órgano de la economía. Una de estas formas es la tuberculosis hepática que tiene cuatro modalidades de presentación: Tuberculoma, abscesos tuberculosos, colangitis tuberculosa y hepatitis granulomatosa tuberculosa (10-13)
La tuberculosis hepática localizada comprende a los tuberculomas y abscesos que cursan con un cuadro general de fiebre, pérdida de peso, anorexia y sobre todo dolor abdominal (14). Hasta 1994 se había reportado 29 casos de abscesos hepáticos tuberculosos en la literatura inglesa y 14 en la japonesa;ello traduce su baja frecuencia, la que alcanza 0. 34% (13)
De otro lado, las enfermedades granulomatosas que cursan con FOD incluyen la sarcoidosis, la colitis ulcerativa, la enfermedad de Crohn, la enfermedad de Whipple y las hepatitis granulomatosas (3). Las causas más frecuentes de hepatitis granulomatosa se listan en la (Tabla 1).
Tabla 1. Hepatitis granulomatasa. Causas.

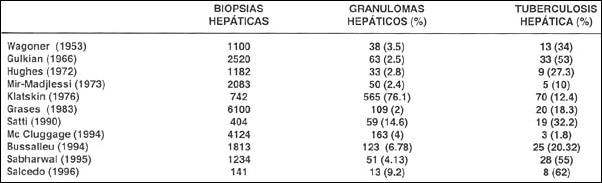
La tuberculosis hepática difusa, miliar o hepatitis granulomatosa tuberculosa se presenta con sintomatología escasa e inespecífica como malestar general, fiebre, escalofríos, anorexia, astenia o pérdida ponderal (12). Se caracteriza por la presencia de granulomas microscópicos en lobulillos hepáticos cercanos a la vena centrolobulillar;y en el 90% de los casos de TBC diseminada existe compromiso hepático (14). En diversas series de granulomas hepáticos la etiología tuberculosa oscila en rangos de 1. 8 a 62% (Tabla 2).
Tabla 2. Granulomatosis hepática tuberculosa.
En nuestro caso el hallazgo que permitió inferir el diagnóstico fue la evidencia de colestasis que como se sabe, con la ecografía o la tomografía axial computarizada se define si es intrahepática o extrahepática. El estudio se complementa con una biopsia hepática en el primer caso, y con una colangiopancreatografía retrógrada endoscópica en el segundo.
El otro evento que ayudó en el diagnóstico fue la disociación fosfatasa alcalina-transaminasas que es un aumento de fosfatasa fuera de proporción en relación con el aumento de la transaminasa glutámica oxalacética. Este hallazgo suele corroborarse con un incremento en el valor de gammaglutamiltransferasa y se ve en pocas entidades, como cirrosis biliar primaria, Fiebre Q, absceso hepático, neoplasia hepática y hepatitis granulomatosa entre otras.
Ciertamente el diagnóstico diferencial de la granulomatosis hepática no es sencillo. En la mayoría de las series alrededor de 50 a 70% de los casos corresponden a sarcoidosis y tuberculosis (14, 15). La diferencia fundamental radica en que en la sarcoidosis los granulomas son más numerosos que en la TBC, siendo necesario en ocasiones repetir la biopsia. Además, en TBC puede o no haber necrosis caseosa, presencia de BAAR y los pocos granulomas tienden a coalescer. En general el 25% de pacientes con TBC pulmonar y el 80% de casos de TBC extrapulmonar tienen hepatitis granulomatosa (16-19).
Como si esto no fuera suficiente el rendimiento de las pruebas para el diagnóstico de tuberculosis hepática son bajas. Así en la serie de Gilinsky de 116 casos el 50% (58 casos) fueron portadores de TBC hepática, se encontraron BAAR en 7 pacientes, necrosis caseosa en 10 y el cultivo fue positivo unicamente en 2 casos. (14).
Un tema de confusión lo constituye la interpretación del examen bioquímico del líquido ascítico en el cual la precisión de estar frente a un exudado o trasudado dependerá de ciertos parámetros de uso clínico como la concentración de proteínas en líquido ascítico cuyo punto de corte es 2.5 g/dl, la gradiente de albúmina con límites en 1.1, y el índice proteico (Proteína ascitis/Proteína sérica) con valores de 50% que muestran especificidades de 73.3, 93.3 y 73.3, y valores predictivos positivos de 85.7, 96.5 y 86.6 respectivamente mostrados en la serie de 40 pacientes de Paré (20,21). Con una casuística de 60 pacientes, Valdivia en su tesis doctoral al modificar los límites de estos parámetros a 2.94 g/dl para la concentración de proteínas en líquido ascítico, a 1. 5 para la concentración de albúmina en líquido ascítico y a 47% para el índice proteico obtiene especificidades de 100, 81.5 y 92.6% y valores predictivos positivos de 100, 85.3 y 94.1 respectivamente. (22-24).
Otro comentario que motivó este caso fue el desarrollo de hiperesplenismo que como se sabe cursa con esplenomegalia, anemia, bicitopenia o pancitopenia, médula ósea normal o con hiperplasia compensadora, reticulocitosis, incremento de macroplaquetas, que se resuelven con la esplenectomía, hallazgos que aquí se observaron casi en su totalidad. Sin embargo llamaba la atención la esplenomegalia masiva que reduce el diagnóstico diferencial a entidades como talasemias, trombosis de la esplénica, hipertensión portal, leucemias mielocíticas y linfocíticas crónicas, leucemia a células velludas, linfoma No Hogkin, tuberculosis, amiloidosis, leishmaniasis visceral, histoplasmosis brucellosis y paludismo crónico.
Creemos que esta paciente tuvo además, esta última condición, llamada también Síndrome de Esplenomegalia Tropical o Esplenomegalia Hiperreactiva Malárica que según los criterios establecidos por Fakunle se caracteriza por: Esplenomegalia masiva y persistente (mayor de 10 cm), residencia prolongada en área malárica, títulos elevados de anticuerpos antimaláricos ( mayor a 2 DE Ig M de la zona) y reducción del tamaño esplénico con profilaxis para malaria (cloroquina 10 mg/Kg/semanal). En estos casos el incremento en el tamaño del hígado y bazo se ven estimulados por la gran cantidad de inmunocomplejos de alto peso molecular formados por Ig M. Se considera que existe malaria crónica en casos de paludismo de larga duración, causados por recaídas o recrudescencias, desencadenados a su vez por: Tratamiento insuficiente, cambios de clima o exposiciones al frio, alteración de la resistencia individual por desnutrición, inmunosupresión, intervenciones quirúrgicas y enfermedades debilitantes. El cuadro clínico característico incluye dolor abdominal, escalofríos, fiebre y sudoración asociadas a anemia, leucopenia y esplenomegalia (25-27).
Se sabe que en algunos casos refractarios a la terapéutica puede aparecer una proliferación linfoide clonal que más tarde puede evolucionar hacia un trastorno linfoproliferativo maligno (27). Nuestra paciente en el seguimiento ha cursado con eventuales síndromes purpúricos asociado a plaquetopenia sin variación del cuadro de hipertensión portal.
En conclusión, la hepatitis granulomatosa es una causa poco frecuente de fiebre de origen oscuro. Su diagnóstico requiere una alta sospecha clínica, en el contexto de colestasis intrahepática con notable incremento de fostasa alcalina que no se acompaña de movilización proporcional de transaminasa oxalacética. En nuestro medio la etiología tuberculosa es la más común, su confirmación depende de los hallazgos anatomopatológicos y la respuesta a la terapia suele ser satisfactoria.
BIBLIOGRAFÍA
1. Arnow PM, Flaherty JP Fiebre de origen desconocido. The Lancet 1998;32(1):50-56.
2. Hirschmann JV Fever of unknown origin in adults. Clin Infect Dis 1997;24:291-302.
3. Gelfand JA, Wolff SM Fiebre de origen desconocido. En Mandell G, Benett J. , Dolin R. Enfermedades Infecciosas, Principios y Práctica. Edit Panamericana 4ta Ed. 1997:589-603.
4. De Kleijn EM, Vandenbroucke JP, Van der Meer JW, et al. Fever of unknown origin I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. Medicine 1997;76(6):392-400.
5. De Kleijn EM, Van Lier HJ, Van der Meer JW, et al. Fever of unknown origin II. Diagnostic procedures in a prospective multicenter study of 167 patients. Medicine 1997;76(6):401-414.
6. Kashiwagi H. Fever of unknown origin: A changing diagnostic spectrum. Internal Med 1994;33(2):65-66.
7. Likuni Y, Okada J, Kondo H, et al. Current fever of unknown origin 1982-1992. Internal Med 1994;33(2):67-74.
8. Ramos JM, Ramos R, Herrero F. Fiebre de origen desconocido en Medicina Interna. Experiencia de autores españoles durante 20 años. Ann Intern Med 1997;14(11):585-592.
9. Luna E. , Carcelen A. , Recavarren S. y col. Tuberculosis hepática. Estudio retrospectivo de 30 pacientes con diagnóstico anatómico en el Hospital General Cayetano Heredia. Rev Gastroent Perú 1983;3:130-135.
10. Fernández-Miranda C., Perpina C., Kessler P. Et al. Hepatosplenic tuberculouos Absccesses in a patient with polyarteritis nodosa. Am J Gastroenterol 1993;88(8):1297-1298.
11. Bandres M., Burstein E., Casas J. Y col. Absceso hepático tuberculoso. Descripción de un caso. Rev Gastroent Perú 1994;14:233-237.
12. Kubota H., Ageta M., Kubo H. Tuberculous liver abscess treated by percutaneous infusion of antituberculous agents. Internal Med 1994;33(6):351-356.
13. Chahud A., Garcia J., Takano J. Y col. Tuberculosis hepática. Estudio prospectivo clínico patológico. Rev Gastroent Perú 1986;6:82-91.
14. Gilinsky NH., Campbell JA., Kirsch R. The clinical spectrum of hepatic granuloma. S Afr Med J 1981;60:691-694.
15. Sartin J., Walker R. Granulomatous hepatitis:A retrospective review of 88 cases at the Mayo Clinic. Mayo Clin Proc 1991;66:914-918.
16. Fauci A., Hofman G. Granulomatous hepatitis. In Principles and Practice of Infectous Disease. Mandell G., Bennett J., Dolin R. Forth Ed Churchill Livingstone 1995;1159-1164.
17. Grases P., Pastran Z., Cáceres A. Y col. Hepatitis granulomatosa. Espectro clínicopatologico en 107 casos con biopsia hepática. GEN 1983;37(3):199-232.
18. Chien RN., Lin PY., Liaw YF. Hepatic tuberculosis: Comparison of miliary and local form. Infection 1995;23(1):5-12.
19. Mc Cluggage WG., Sloan JM. Hepatic granulomas in Northern Ireland: A thirteen year review. Histopathology 1994;25:219-228.
20. Lenoir P., Glibert L., Vandenplas A., et al. Tuberculous peritonitis in an adolescent male. Europ J Gastroent Hepatol 1995;7:477-480.
21. Martinez-Vasquez JM., Ocaña I., Ribera E., et al Adenosine deaminase activity in the diagnosis of tuberculous peritonitis. Gut 1986;27:1049-1053.
22. Bussalleu A., Clendenes D., Berriós J., y col. La biopsia hepática en el Hospital Nacional Cayetano Heredia. Rev Med Hered 1994;5(3):129-137.
23. Salcedo R., Valdivia M., Zapata C. La laparoscopía como método diagnóstico clínico en un centro hospitalario del Perú. Rev Gastroent Perú 1996;16:34-38.
24. Valdivia M. La validez de la concentración de proteínas en el líquido ascítico y suero para el diagnóstico diferencial de las ascitis. Tesis Doctoral. Universidad Peruana Cayetano Heredia. Lima-Perú. 1996.
25. Alegrim WD., Alegrim MG., Albuquerque BC., et al. Esplenomegalia tropical no Rio Ituxi, Amazonas, Brasil. Rev Inst Med Trop Sao Paulo 1982;Supl 6(6):54-57.
26. Espinal C., Arias A., Rodríguez C y col. Síndrome de esplenomegalia tropical: Presentación del primer caso confirmado en Colombia:Biomédica 1983;3(1-2):26-30.
27. Wallace S., Bedu-Addo G., Rutherford TR., et al. Serological similarities between hyperreactive malarial splenomegaly and splenic lymphoma in West Africa. Trans Roy Soc Trop Med Hygiene 1998;92:463-467.