REPORTE DE CASOS CLÍNICOS
Hemocromatosis Hereditaria
Lidia Landa Garrido *, José Huamán Muñante **, Víctor Valencia Caballero **, Zenaida Lozano Miranda ***, Augusto Nago Nago ****
*Médico Cirujano, Servicio de Medicina del Policlínico San Antonio de Padua Lima-Perú
** Médico Internista Asistente del Hospital Arzobispo Loayza Lima-Perú
*** Médico Anatomopatólogo Asistente del Hospital Arzobispo Loayza Lima-Perú
**** Médico Gastroenterólogo Asistente del Hospital Arzobispo Loayza Lima-Perú
RESUMEN
Se presenta el caso de un varón de 43 años, con diagnóstico de diabetes
mellitus, insuficiencia cardiaca, pigmentación de piel, cirrosis
hepática, y de hemocromatosis hereditaria confirmada por biopsia
hepática. El objetivo de esta publicación es tener presente como
diagnóstico diferencial a la hemocromatosis ante un paciente en la edad
media de la vida con varias patologías y órganos involucrados.
Palabras Clave: Hemocromatosis, ferritina sérica, sobrecarga férrica.
SUMMARY
The case of a 43-year-old male is presented, with diagnosed diabetes
mellitus, heart failure, skin pigmentation, hepatic cirrhosis, and
hereditary hemochromatosis confirmed by liver biopsy. The objective of
this publication is to have hemochromatosis in mind as a differential
diagnosis in a middle-aged patient with several pathologies and organs
involved.
Key Words: Hemochromatosis, serum ferritin, iron overload
INTRODUCCIÓN
La hemocromatosis es una enfermedad que se caracteriza por la
sobrecarga férrica progresiva en diversos órganos. Existen dos tipos:
la primera, idiopática, hereditaria o genética y la secundaria
o hemosiderosis.(1,2,3,4) La primera descripción clínica de
glucosuria, cirrosis e hiperpigmentación de la piel fue hecha en 1865,
por Trousseau. En 1889. Reckinghausen es el primero en usar el término
de hemocromatosis. Ya en 1976 Simon y colaboradores demostraron que el
gen se encontraba en la región HLA del cromosoma 6, siendo transmitida
por herencia autosómica recesiva que en la mayoría de los pacientes es
causada por mutación en el gen HFE, que se localiza en el brazo corto
del cromosoma 6 (1,2,3,4). Se han identificado dos mutaciones del gen
HFE en estos pacientes: la C282Y (sustitución de cisteína por tirosina
en la posición 282) y la H63D (sustitución de histidina por aspartato en
el aminoácido 63). Casi el 90% de los pacientes son homocigotos
C282Y/C282Y y un 5% heterocigoto C282Y/H63D y el 5% restante no se
asocia a mutación del gen HFE (1,2,3,5,6,7,8,12,13).
Los homocigotos de C282Y tienen un riesgo más alto para la sobrecarga de
hierro dependiendo de la presencia probable de otros modificadores,
genéticos o medio ambientales, que contribuye a la expresión clínica de
hemocromatosis (9). La prevalencia en la población blanca es más común,
con una frecuencia estimada en homocigotos de 1:220 a 1:400(3). La
proporción entre hombres y mujeres es de 8:1 a 2:1 (3). La
Hemocromatosis es de inicio temprano en varones y más severa que en las
mujeres, por las pérdidas fisiológicas de hierro durante la menstruación
y el embarazo. Los síntomas ocurren mayormente en la cuarta década,
reportándose también en pacientes jóvenes.
El gen HFE
codifica la síntesis de una proteína transmembrana, que se une al
receptor de la transferrina, la distribución tisular de la proteína HFE
es amplia (10), la consecuencia de la mutación C282Y es la ausencia de
expresión de la proteína HFE en la superficie de la célula, mientras que
la mutación H63D es la alteración de la estructura terciaria de está
proteína, resultado de esto es la absorción excesiva de hierro por la
mucosa duodenal acumulándose progresivamente en las células epiteliales
del hígado, hipófisis, páncreas y músculo cardiaco, provocando fibrosis e
insuficiencia funcional (1,2,3,4).
Los síntomas
tempranos son inespecíficos, incluyen fatiga, artralgia, disfunción
eréctil, e incremento de la pigmentación de la piel. Al progresar la
enfermedad se desarrolla hepatomegalia, fibrosis hepática y cirrosis hay
un incremento en la incidencia de carcinoma hepatocelular. Los
depósitos de hierro en el corazón causan cardiopatía usualmente
congestiva pero puede ser restrictiva o asociada con pericarditis y
arritmias. La asociación a endocrinopatías incluyen diabetes mellitus,
hipopituitarismo, hipogonadismo e hipoparatiroidismo. Los pacientes con
hemocromatosis son más susceptibles a infección particularmente con
Vibrio vulnificus, Listeria monocytogenes, Yersinia enterocolítica,
Salmonella enteriditis serotipo typhimurium, Klebsiella pneumoniae,
Escherichia coli, Rhizopus arrhizus y especies mucor(1,4).
La hemocromatosis secundaria se presenta en pacientes con anemia
crónica y que han recibido transfusiones múltiples, otras causas menos
comunes son déficit congénito de transferrina, ingesta excesiva de
hierro (siderosis Bantu), porfiria cutánea tarda y como complicación de
la derivación porto cava(1,2,3,4). En los casos de sobrecarga secundaria
los depósitos de hierro se encuentran primero en las células de Kupffer
y posteriormente en los hepatocitos. (23)
CASO CLÍNICO
Paciente varón, de 43 años de edad, natural de Huarmaca, provincia de
Huancabamba, departamento de Piura, procedente de Lima, con una estancia
mayor de seis meses, casado, raza mestiza, de ocupación mecánico.
Antecedente de consumo de alcohol ± 100g a la semana
desde los 10 años, fiebre tifoidea a los 20 años. Padre del paciente
fallecido por cirrosis, diez años antes le diagnosticaron diabetes
mellitus. Tía materna con diabetes mellitus, y tío fallecido por infarto
del miocardio.
Acude al servicio de medicina de el
Hospital Loayza el 08/11/01, con un tiempo de enfermedad de 1 año,
inicia síntomas con astenia, fatiga, disminución de la libido por
disfunción eréctil, pérdida de peso de ± 10 Kg en los últimos seis meses
y calambres por las noches. Siete meses antes de su ingreso presentó
polidipsia, poliuria y polifagia, por lo que acudió a un establecimiento
de salud donde le diagnosticaron diabetes mellitus e inician
tratamiento con glimepirida 4mg/d, además le realizaron una endoscopía
por presentar dispepsia y epigastralgia diagnosticándole gastritis.
Desde hace 4 meses nota edema de miembros inferiores, disnea progresiva y
palpitaciones, por lo cual acude al Hospital Nacional Arzobispo Loayza,
notando además hiperpigmentación de cara y miembros superiores. Siendo
hospitalizado para completar estudio diagnóstico.
Al
examen físico se encuentra paciente en aparente regular estado general,
nutrición y de hidratación. FC: 76X' FR: 16X' °T: 36,8 °C PA: 130/80
mmHg. Piel y faneras : Pigmentación negruzca, brillante de aspecto
bronceado en cara y miembros superiores (foto 1).
Vello axilar y pubiano disminuido. TCSC: Edema en miembros inferiores
hasta rodilla. Cardiovascular: Ruidos cardiacos arrítmicos, de buena
intensidad, no soplos. Abdomen: Blando, depresible, ruidos hidroaéreos
presentes, span hepático ± 14 cm, se palpa a 3 cm debajo del reborde
costal derecho. Bazo palpable, punta a 3 cm debajo del reborde costal
izquierdo, no doloroso a la palpación.
EXAMENES AUXILIARES:
Hb: 13,2mg/%, Glucosa: 159 - 351mg/dl (4 mediciones), Hemoglobina
glicosilada: 7.65% (VN: 4.2 a 6 %), Creatinina: 0.64mg/dl, VSG: 24mm/h
Colesterol: 182mg/dl, Triglicéridos: 108mg/dl, TGP: 73-113U/L (2
mediciones), TGO: 86 - 110U/L (2 mediciones), FA: 321-393U/L (2
mediciones) , Bilirrubina total: 1.7mg/dl, BD:0.48, BI:1.3, TP:10.8",
Proteínas total: 7.5 g/dl , Albúmina: 5g/dl, Proteinuria:0.187g/L.
AgHBs, VHC, AgHBe y AntieVHB, no reactivos.
Eco abdominal: Hepatoesplenomegalia difusa homogénea.
Endoscopía: Gastritis crónica superficial y atrófica con atipia leve a moderada, metaplasia intestinal.
Ecocardiograma: Dilatación de los cuatro ventrículos a predominio
derecho, severa hipocinesia difusa del ventrículo izquierdo. Fracción de
eyección sistólica 45%. Aumento de ecos en pared ventricular izquierda.
Hierro sérico: 278ug/dl (VN: 59-158), ferritina:
1102.5ng/ml (VN: 30-400), transferrina: 255.81mg/dl(VN: 200-400),
Saturación de transferrina: 108.6% (VN: 25-50%).
Alfafetoproteína: 2.01ng/ml(2-10ng/ml)
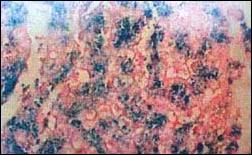
Biopsia hepática: Parénquima hepático con presencia de depósito de
hierro intracelular, con distribución pericanalicular granular (foto 2 y 3).
Foto1. Pigmentación negruzca, brillante de piel

Foto 2. Biopsia hepática (Hematoxilina y eosina, 400X),
presenta depósitos de pigmentación marrón
a nivel intracelular y pericanalicular.

Foto 3. (Perlas de azul de prusia, 400x), detecta la presencia
de depósitos de hierro en parénquima hepático con distribución
pericanalicular granular intracelular, característico de
Hemocromatosis, grado histológico +3.
DISCUSIÓN
El presente caso clínico es de un hombre de 43 años, que inicia su
enfermedad hace un año con astenia, fatiga, y disfunción eréctil.
Niederau et al reportaron debilidad o letargia en un 83% de un total de
163 pacientes, así como pérdida de la líbido e impotencia en un 38%,
pigmentación de piel (75%), diabetes (55%), hepatomegalia (83%),
cirrosis 69% y en los exámenes de laboratorio: enzimas hepáticas
anormales (62%), las cuáles presentó el paciente, reportándose también
en la literatura dolor abdominal 58% y artralgias 43% las cuales no
estuvieron presentes en el paciente; Adams et al y Bacon y Sadiq(3,14),
mencionan incidencia de diabetes en un 11 y 5% respectivamente.
El diagnóstico de hemocromatosis se establece
confirmando las alteraciones del metabolismo del hierro, primero por el
indice de saturación de transferrina por encima del 50% en mujeres y
mayor del 62% en varones siendo el indicador analítico más sensible, su
determinación se hace en ayunas. La ferritina sérica mayor de 300ng/ml
en varones y por encima de 200ng/ml en mujeres también se observa en
hemocromatosis hereditaria con transferrina normal y nivel sérico de
hierro elevado entre 180-300ug/dl; (1,2,3,4,20,21) siendo criterios
diagnósticos para hemocromatosis hereditaria. Sin embargo la ferritina
no es un indicador sensitivo puede estar muy incrementada en diversos
procesos inflamatorios(17,18,19) no obstante se debe descartar cirrosis
ante un paciente con hemocromatosis con ferritina superior a
1000ng/ml.
En el paciente encontramos un hierro
sérico elevado (278ug/dl), una ferritina de 1102.5ng/ml, saturación de
transferrina muy incrementada (108.6%) y una transferrina normal.
Descartándose otras causas de sobrecarga férrica, se decide hacer la
biopsia hepática para la localización, cuantía del depósito de hierro y
grado de lesión histológica, realizándose la biopsia hepática
percutánea ayudándose de la coloración perla azul de prusia, donde se
encontró depósitos de hierro a nivel intracelular, con un patrón
pericanalicular granular, con un grado de lesión histológica de +3, no
pudiendo definir fibrosis porque la muestra no contenía espacio porta,
pero por el grado de lesión(1,3) se confirma la presencia de cirrosis,
concluyendo que el paciente presenta una hemocromatosis hereditaria con
una cirrosis hepática, la ingesta de alcohol puede haber incrementado la
absorción de hierro, además tiene el antecedente de un cuadro semejante
en su padre.
Al paciente se le realizó una
flebotomía semanal de 500ml, equivalente a 250mg de hierro, siendo
transferido a Hospital de ESSALUD, para continuar con flebotomías, las
cuales deben realizarse en forma semanal hasta que la hemoglobina
alcance 11g/dl o permanezca con un hematocrito debajo de 37%, cada 2 ó 3
meses evaluar saturación de transferrina y ferritina, una vez que los
depósitos de hierro estén depletados (ferritina < 50ng/ml; saturación
de transferrina < 50%) se procede a mantener la flebotomía de una
unidad cada 2 ó 3 meses(3) y además se debe hacer control ecográfico
abdominal y alfafetoproteina sérica cada 6 meses en los pacientes con
cirrosis(1,2,3,4,24) para monitorizar al paciente ante un potencial
desarrollo de carcinoma hepatocelular cuyo riesgo es mayor en
homocigotos para la mutación HFE(15,22) ocurriendo en un 30% en
pacientes con cirrosis(1). Es necesario hacer un despistaje familiar en
parientes de primer y segundo grado; dosar saturación de transferrina y
ferritina si estos resultados son elevados se procede a una biopsia
hepática para histoquímica y determinación bioquímica del contenido de
hierro, complementando con estudio genético. En el caso de nuestro
paciente tiene dos hijos varones de 20 años y de 16 años, presentando
uno de ellos ya pruebas alteradas del metabolismo del hierro estando al
momento asintomático, lo cual indicaría una evaluación más exhaustiva
del descarte de hemocromatosis.
BIBLIOGRAFíA
1. DANTAS W. Hemocromatose Hereditária. Rev Gastroenterol Perú 2001;21:42-55
2. DE CASO A, LÓPEZ-HUERTA M, TERRAZO M y cols. Gen de la Hemocromatosis primaria: implicaciones diagnósticas y terapéuticas. Medicina General 2000;29:970-975
3. BACON B AND BRITTON R. Hereditary Hemochro-matosis. Gastrointestinal and Liver disease. 6th Edition, 1998;2:1097-1103
4. ANDREWS N. Disorders of Iron Metabolism. N Engl J Med 1999;341:1986-1995 (book review)
5. PIETRANGELO A, MONTOSI G, TOTARO A, et al. Hereditary Hemochromatosis in Adults without Pathogenic Mutations in the Hemochromatosis Gene. N Engl J Med 1999;341:725-732
6. PIETRANGELO A. Hemochromatosis: Genetics, pathophysiology, diagnosis and treatment. N Engl J Med 2000;343:516-517
7. BURKE W, IMPERATORE G, MCDONNELL SM, et al. Contribution of different HFE genotypes to iron overload disease: a pooled analysis. Genetics in Medicine 2000;2:271-277
8. WAHEED A, PARKKILA S, ZHOU XY, et al. Hereditary hemochromatosis: Effects of C282Y and H63D mutations on association with 2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci 1997;94:12384-89
9.- BULAJ Z, AJIOKA R, PHILLIPS J, et al. Disease-Related Conditions in Relatives of Patients with Hemochromatosis N Engl J Med 2000;343:1529-1535
10. WAHEED A, PARKKILA S, SAARNIO J. Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum. Proc Natl Acad Sci 1999;96:1579-84
11. SCULLY R, MARK E, MCNEELY W, et al. Case Records of the Massachusetts General Hospital N Engl J Med
12. ADAMS PC. Population screening for haemochro-matosis. Gut 2000; 46: 301-303
13. EDWARDS CQ, GRIFFEN LM, GOLDGAR D, et al. Prevalence of hemochromatosis among 11,065 presumably healthy blood donors. N Engl J Med 1988;318:1355-62
14. GORE J AND FALLON J. Case 31-1994 A 25-Year-Old Man with the Recent Onset of Diabetes Mellitus and Congestive Heart Failure. N Engl J Med 1994; 331: 460-466
15. WILLIS G, WIMPERIS JZ, LONSDALE R, et al. Incidence of liver disease in people with HFE mutations. Gut 2000;46:401-404
16. TUOMAINEN TP, KONTULA K, NYYSSÖNEN K, et al. Increased Risk of Acute Myocardial Infarction in Carriers of the Hemochromatosis Gene Cys282Tyr Mutation.Circulation 1999;100:1274-1279
17. CIFUENTES P, DOWLING P AND OZAKI R. ¿Should all patients with diabetes mellitus be screened for hemochromatosis?. West J Med 2002;176:110-114
18. FORD ES AND COGSWELL ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 1999;22:1978-1983
19. PÉREZ DE NANCLARES G, CASTAÑO L, GAZTAMBIDE S, et al. Excess Iron Storage in Patients with Type 2 Diabetes Unrelated to Primary Hemochromatosis. N Engl J Med 2000;343:891
20. EDWARDS C, KUSHNER J. Screening for Hemochromatosis. N Engl J Med 1993;328:1616-1620
21. POWELL L, GEORGE K, MCDONNELL, S, et al. Diagnosis of Hemochromatosis. Ann Intern Med 1998; 129:925-931
22. BACON B, OLYNYK J, BRUNT E, et al. HFE Genotype in patients with Hemochromatosis and other Liver diseases. Ann Intern Med 1999;130:953-962
23. BRAVO A, SHETH S AND CHOPRA S. LIVER BIOPSY. N Engl J Med 2001;344:495-500
24. BARTON J, MCDONELL S, ADAMS P, et al. Management of Hemochromatosis. Ann Intern Med 1998;129:932-939